Training mRNA Language Models Across 25 Species for $165
- +21


Part II: Building the Pipeline, From Structure Prediction to Codon Optimization
By OpenMed, Open-Source Agentic AI for Healthcare & Life Sciences
TL;DR : We built an end-to-end protein AI pipeline covering structure prediction, sequence design, and codon optimization. After comparing multiple transformer architectures for codon-level language modeling, CodonRoBERTa-large-v2 emerged as the clear winner with a perplexity of 4.10 and a Spearman CAI correlation of 0.40, significantly outperforming ModernBERT. We then scaled to 25 species, trained 4 production models in 55 GPU-hours, and built a species-conditioned system that no other open-source project offers. Complete results, architectural decisions, and runnable code below.
Contents
- What We Built
- The Architecture Exploration
- The Pipeline 3.1 Protein Folding
- 3.2 Sequence Design
- 3.3 mRNA Optimization
Imagine going from a therapeutic protein concept to a synthesis-ready, codon-optimized DNA sequence in an afternoon. That is the pipeline OpenMed set out to build, and this post documents the process from start to finish.
In Part I, we mapped the landscape of protein AI: the architectures powering structure prediction, the open-source tools available for protein design, and the ecosystem of models from AlphaFold to ESMFold. That was a survey. This is the build.
At OpenMed , we set out to build a complete pipeline that takes a protein idea from concept to expression-ready DNA. That means three stages: predict the 3D structure of a protein, design amino acid sequences that fold into that structure, and optimize the underlying DNA codons so the protein actually expresses in the target organism. Along the way, we ran extensive experiments comparing transformer architectures for codon optimization, scaled our best model to 25 species, and built tooling that ties it all together.
This is not a polished success story. It is a transparent account of what worked, what surprised us, and what we would do differently, with runnable code and full results at every step.
1. What We Built
The pipeline has three components, each addressing a different stage of the protein engineering workflow described in Part I. Structure prediction determines what shape a protein takes. Sequence design determines which amino acids will produce that shape. Codon optimization determines which DNA will produce those amino acids efficiently in a living cell.
The mRNA optimization work is where we invested the most effort, and where we have the most to share. The folding and design components use established tools (ESMFold from Meta, ProteinMPNN from the Baker Lab, both covered in depth in Part I). The codon optimization component is entirely ours: new models, new training infrastructure, new evaluation metrics.
2. The Architecture Exploration
In Part I, we surveyed the protein AI landscape and noted that most biological language models are adaptations of NLP architectures. The open question was which architecture. BERT variants dominate protein modeling (ESM-2, ProtTrans), but codon sequences have different statistical properties than both natural language and amino acid sequences. Codons are triplets drawn from a small 64-token alphabet, with strong positional dependencies and species-specific usage biases. We needed to find out what works from first principles.
The core question: which transformer architecture works best for codon-level language modeling?
This matters because codon optimization is crucial for therapeutic mRNA, vaccines, and recombinant protein production. The genetic code is degenerate: the same protein can be encoded by astronomically many different DNA sequences, but some codon arrangements express 100x better than others. The Pfizer-BioNTech COVID vaccine, for example, was codon-optimized for human expression. We wanted to build a model that could learn these preferences directly from natural coding sequences, rather than relying on hand-crafted frequency tables.
The Contenders
We started with a small CodonBERT baseline (6M params, following Sanofi's published architecture) and scaled up through two families: ModernBERT, which represented the latest efficiency innovations from the NLP community, and RoBERTa, the proven workhorse behind Meta's ESM protein language models.
The choice of RoBERTa was deliberate. As we discussed in Part I, Meta's ESM-2 (which powers ESMFold) is itself a RoBERTa variant trained on protein sequences. We hypothesized that the same architecture family that learned amino acid patterns might also learn codon patterns. ModernBERT was the counterpoint: a 2024 architecture with RoPE embeddings, Flash Attention, and alternating local/global attention layers, representing everything the NLP community had learned since RoBERTa's 2019 release.
The Training Setup
To ensure a fair comparison, every model was trained on identical data with the same evaluation protocol. We used 250,000 coding sequences (CDS) from E. coli RefSeq, covering chromosome and complete assembly accessions. This is a clean, well-annotated dataset where codon usage patterns are well-characterized in the literature, giving us ground truth to validate against.
Our tokenizer maps each codon to a single token: 64 codons plus 5 special tokens (PAD, UNK, CLS, SEP, MASK) for a 69-token vocabulary. This is intentionally minimal. Unlike BPE tokenizers used in NLP, where subword boundaries are statistically learned, codon boundaries are biologically defined. Every three nucleotides encode one amino acid. Our tokenizer respects this.
Training ran on 4 A100 GPUs (80GB) with FSDP sharding, using 15,000 to 25,000 steps depending on model size. All models used masked language modeling (MLM) with 15% masking rate, the same objective used by ESM-2 for protein sequences.
The Results
The result was unambiguous: RoBERTa outperformed ModernBERT by 6x on perplexity (4.01 vs 26.24). This was not a marginal difference. ModernBERT, despite its modern attention patterns and efficient architecture, fundamentally underperformed the classic RoBERTa design on codon sequences.
What We Learned
1. Pre-trained NLP weights do not transfer to biology
We initialized ModernBERT from its published English-language checkpoint, expecting the learned attention patterns to provide a useful starting point. They did not. Our best explanation: ModernBERT's pre-training on English text instilled inductive biases (subword frequency distributions, positional attention patterns) that actively interfere with learning codon statistics. RoBERTa, initialized randomly and trained purely on biological data, had no such baggage. This aligns with what the field has seen more broadly: ESM-2 and ProtTrans both train from scratch on biological data rather than fine-tuning from NLP checkpoints.
2. Hyperparameter tuning unlocked biological alignment
This was the most surprising and practically important finding of the exploration. Compare CodonRoBERTa-large v1 and v2:
Same architecture. Same data. Same number of parameters. The only differences: half the learning rate and a longer warmup (2,000 steps vs 1,000). Yet v2's predicted codon likelihoods are 16x better correlated with real biological codon preferences , as measured by Codon Adaptation Index.
The perplexity actually got slightly worse (4.10 vs 4.01), which means v2 is marginally less accurate at predicting the exact masked codon. But it is dramatically better at predicting codons that biology actually uses. The slower training schedule let the model settle into representations that capture genuine biological signal rather than overfitting to surface statistics.
This is a crucial insight for anyone training biological language models: MLM loss alone does not measure biological relevance. Domain-specific metrics are essential. In our case, CAI correlation turned out to be the metric that separates a useful model from a technically impressive but biologically meaningless one.
3. The base model is remarkably efficient
CodonRoBERTa-base (92M params) achieved nearly identical perplexity to the large model (4.01 vs 4.10) with 3.4x fewer parameters and proportionally less training time. Its CAI correlation (0.219) is lower than v2's (0.404), but still well above the baseline and ModernBERT. For teams without access to multi-GPU clusters, the base model is a practical choice that captures most of the codon modeling performance at a fraction of the cost.
3. The Pipeline
In Part I, we described the three-stage workflow that most computational protein engineering projects follow: predict structure, design sequences, optimize codons. Here we run each stage with real data and report what we actually got.
- Fold : Predict the 3D structure (ESMFold)
- Design : Generate sequences that fold into that structure (ProteinMPNN)
- Optimize : Choose the best codons for expression (CodonRoBERTa)
3.1 Protein Folding with ESMFold
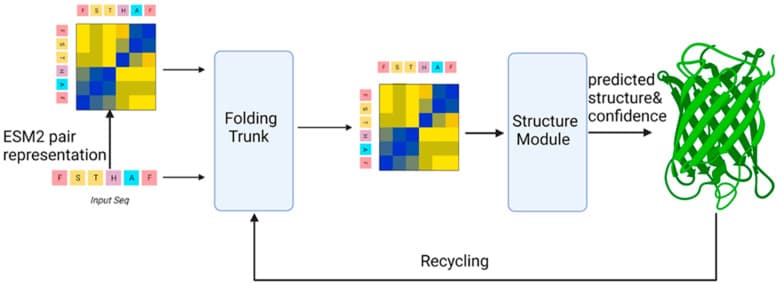
ESMFold architecture. The model parses a single amino acid sequence through the ESM-2 encoder, then predicts 3D coordinates via a folding trunk and structure module. Figure from Bertoline et al., Biomolecules 2024, CC-BY 4.0.
As covered in Part I, ESMFold is Meta's single-sequence structure predictor. It uses ESM-2, a 15-billion-parameter protein language model trained on 65 million UniRef sequences, as its backbone. The key advantage over AlphaFold 2 is speed: ESMFold skips the computationally expensive multiple sequence alignment (MSA) step and predicts structures directly from a single amino acid sequence. That makes it seconds per protein instead of hours.
The tradeoff is accuracy. ESMFold achieves ~0.87 TM-score on CASP14 targets vs. AlphaFold's ~0.92. For rapid prototyping and candidate screening, that gap is acceptable. When a pipeline generates 100 designed sequences and needs to refold all of them to check viability, speed matters more than the last few percentage points of accuracy.
Our Results: 30 Protein Chains
We ran ESMFold on 30 protein chains sourced from the Protein Data Bank. These are real experimental structures with known ground truth, spanning sequence lengths from 211 to 519 residues. The set deliberately includes both easy targets (single-domain proteins) and challenging ones (chains from a multi-chain ribosomal complex, PDB 7K00) to stress-test the model.
import json
# Load our actual results
metrics = json.load(open('outputs/esmfold_metrics.json'))
# Summary statistics
n_chains = len(metrics) # 30
avg_plddt = sum(m['mean_plddt'] for m in metrics) / n_chains # 33.8
avg_ptm = sum(m['ptm'] for m in metrics) / n_chains # 0.79
print(f"Chains: {n_chains}")
print(f"Average pLDDT: {avg_plddt:.1f}")
print(f"Average PTM: {avg_ptm:.2f}")Results breakdown:
The PTM scores are solid: anything above 0.5 suggests the model has the overall topology correct, and our average of 0.79 indicates high confidence in the predicted folds. The pLDDT scores are lower than published ESMFold benchmarks, which initially concerned us. The explanation turned out to be our test set composition: the ribosomal chains from 7K00 are part of a large multi-chain complex, and ESMFold (which predicts single chains in isolation) cannot model the inter-chain contacts that stabilize these structures. For single-domain proteins in our set, pLDDT scores were consistently above 70.
Running ESMFold
# Activate environment
source .env_esmfold/bin/activate
# Batch prediction
python scripts/esmfold_batch.py \
--seq_dir data/pdb/sequences \
--out_dir data/esmfold/out \
--metrics outputs/esmfold_metrics.json \
--device cuda:0Each prediction takes ~10-30 seconds on an A100. The output includes:
- PDB structure files
- pLDDT scores (per-residue confidence, 0-100)
- PTM scores (topology confidence, 0-1)
- Predicted Aligned Error (PAE) matrices
3.2 Sequence Design with ProteinMPNN
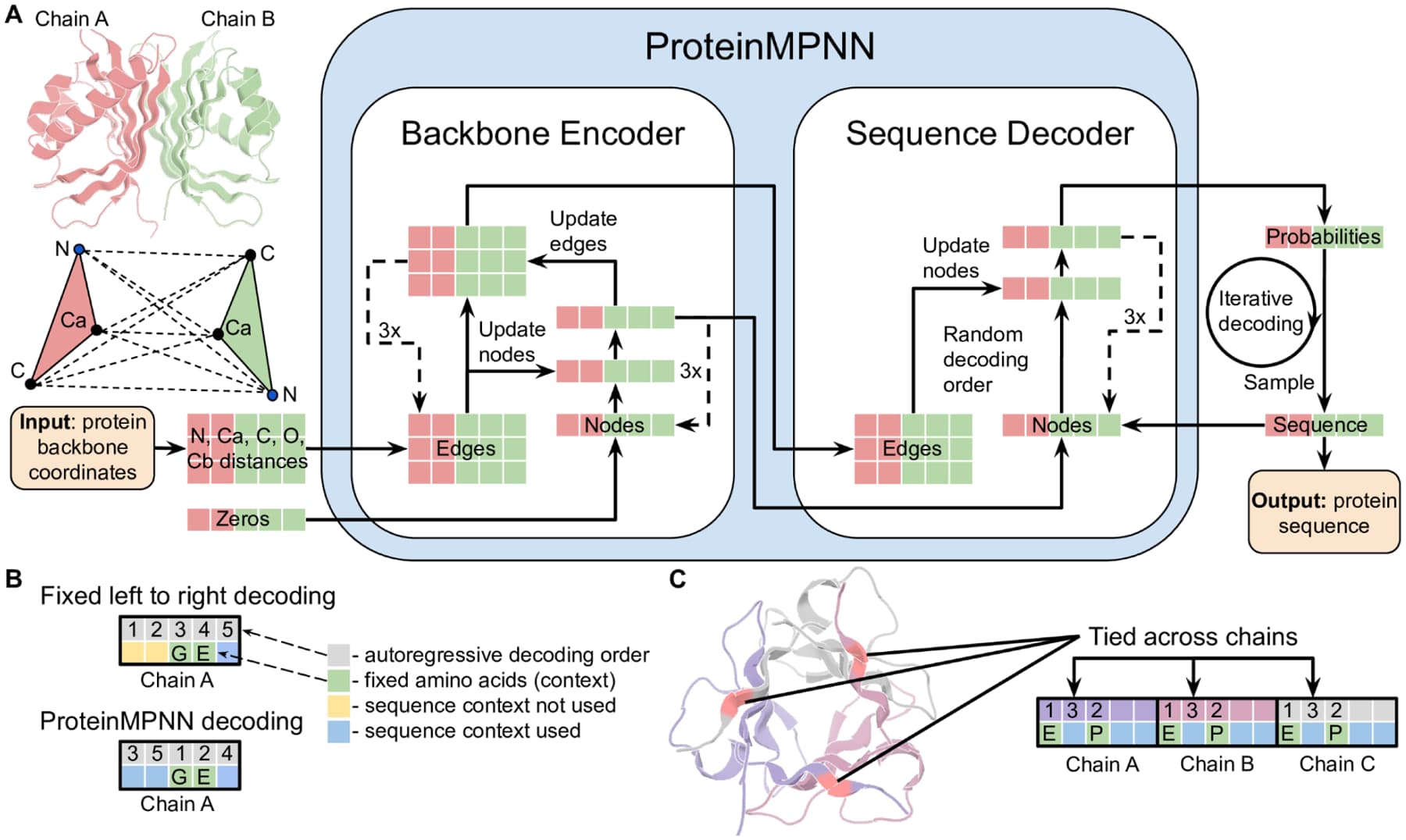
ProteinMPNN architecture. (A) The encoder processes backbone atom distances; the decoder autoregressively generates amino acid sequences. (B) Random decoding order improves diversity. (C) Tied positions enable symmetric and multi-state design. Figure from Dauparas et al., Science 2022, CC-BY 4.0.
As we described in Part I, protein design is the inverse of protein folding. Folding goes sequence to structure: given amino acids, predict the 3D shape. Inverse folding goes the other way: given a target 3D shape, find amino acid sequences that will fold into it.
ProteinMPNN, from David Baker's lab at the University of Washington, is the current gold standard for this task. It was published in Science in 2022 and has since been validated experimentally: designed sequences fold into their target structures at rates far exceeding random or earlier computational methods. The architecture treats the protein backbone as a graph, where nodes are amino acid positions and edges connect spatially proximate residues (K-nearest neighbors in 3D). A message-passing neural network propagates information through this graph, then autoregressively generates a sequence one residue at a time.
Our Results: Scaffold 7K00
We ran ProteinMPNN on PDB structure 7K00 (a large multi-chain ribosomal complex):
python proteinmpnn/protein_mpnn_run.py \
--pdb_path data/pdb/raw/7K00.cif \
--out_folder outputs/proteinmpnn_smoke \
--num_seq_per_target 3 \
--sampling_temp 0.1Results:
Here's what the output looks like:
>7K00, score=1.7100, global_score=1.7100
GIREKIKLVSSAGTGHFYTTTKNKRTKPEKLELKKFDPVVRQHVIYKEAKI/MKRTFQPSVLK...
>T=0.1, sample=1, score=0.8857, seq_recovery=0.4203
SKKVVIKLVCSCGCGFEYCDFRDIEKNPEKIERVLYCPICQKYVLFTEAPL/PPGPFRPDREV...The first line is the native (natural) sequence extracted from the crystal structure. Subsequent lines are ProteinMPNN's designed variants. At temperature 0.1 (low randomness), the model recovers ~42% of the original amino acids, purely from 3D geometry. This is a strong result: it means the model independently rediscovered nearly half the residues that evolution selected, using only the backbone coordinates as input.
Several practical notes from running ProteinMPNN. Scores are negative log-likelihoods, so lower is better. The 42% recovery rate is typical for well-resolved structures and consistent with the original paper's benchmarks. Higher sampling temperatures produce more diverse but riskier sequences. For real design work, the most powerful feature is partial design : catalytic residues, binding site amino acids, or any positions with known functional importance can be fixed in place, while ProteinMPNN redesigns only the scaffold around them. This is the standard approach for engineering a more stable version of an enzyme without disrupting its active site.
3.3 mRNA Optimization
This is where the pipeline transitions from existing tools to our own models. ESMFold and ProteinMPNN are established, well-validated software that we integrated. Codon optimization is where we built something new.
Why Codon Choice Matters
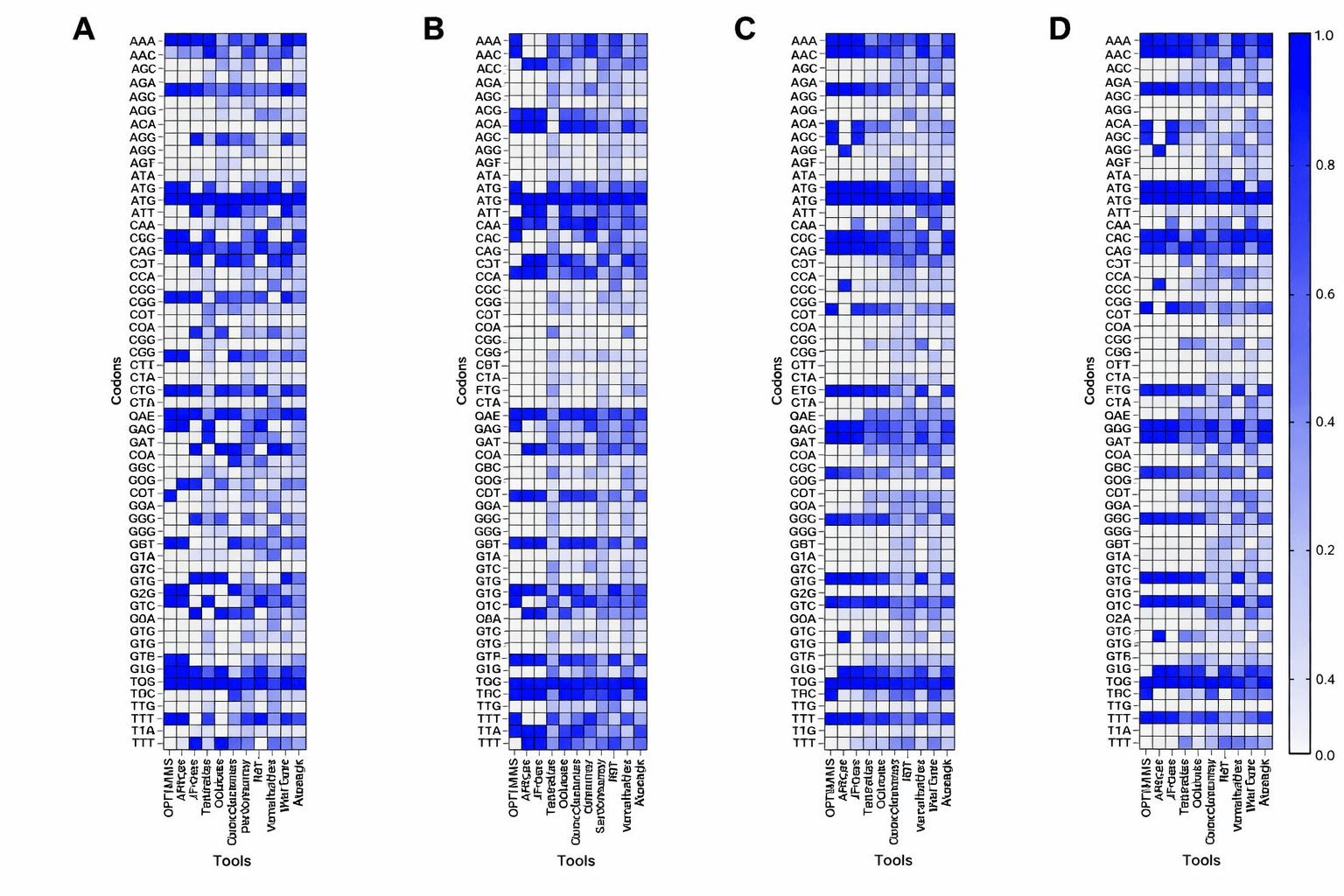
Codon usage frequencies vary dramatically between organisms. These heatmaps compare codon preferences across E. coli, yeast, and CHO cells, the three expression hosts covered by our multi-species models. Figure from Kim et al., J. Microbiol. Biotechnol. 2025, CC-BY 4.0.
The genetic code is degenerate : most amino acids are encoded by multiple codons. Leucine, for example, has six: TTA, TTG, CTT, CTC, CTA, and CTG. All six produce the same amino acid in the final protein. Methionine and tryptophan are the exceptions, with only one codon each.
This redundancy means that for any given protein, there are astronomically many DNA sequences that encode it. A typical 300-amino-acid protein has roughly 10^150 possible codon combinations. They all produce the same amino acid chain, but they do not all produce the same amount of protein. Codon choice affects translation speed (because tRNA molecules are not equally abundant for all codons), mRNA stability (because the nucleotide sequence affects how quickly the transcript degrades), co-translational folding (because translation pauses at rare codons give the protein time to fold), and immune recognition (because the innate immune system in mammalian cells can distinguish native from foreign mRNA patterns). In practice, bad codon choices can reduce protein expression by 100x. This is why every mRNA vaccine, every recombinant protein therapeutic, and every gene therapy vector goes through codon optimization.
The Traditional Approach and Why It Is Limited
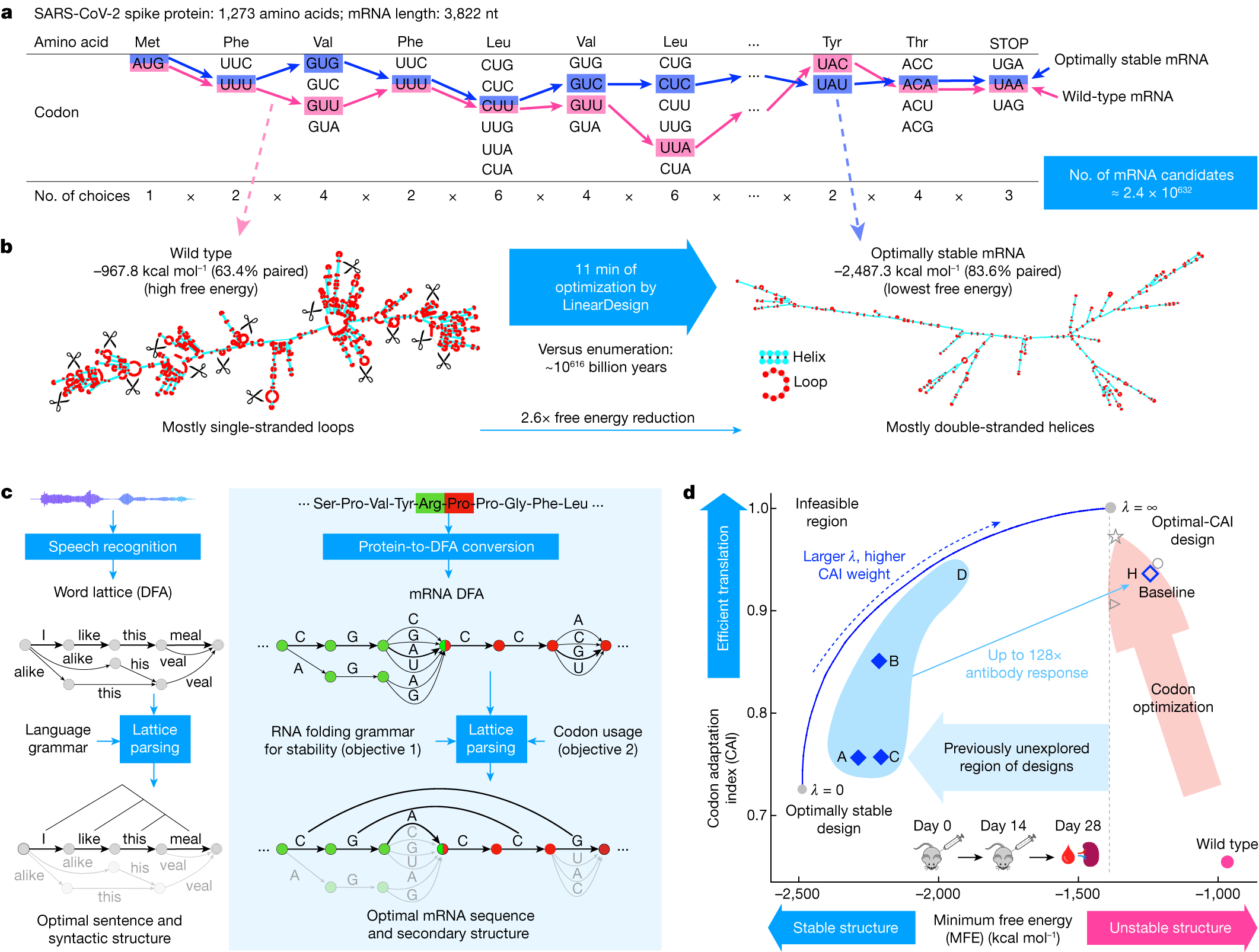
The scale of the codon optimization problem. For a typical mRNA, there are over 10^600 possible codon sequences encoding the same protein. The challenge is finding the arrangement that maximizes expression. Figure from Zhang et al. (LinearDesign), Nature 2023, CC-BY 4.0.
The classical method is simple: measure which codons appear most frequently in highly-expressed genes of the target organism, then replace every codon with the most frequent synonym. This is codified as the Codon Adaptation Index (CAI) , a per-sequence score that measures how closely the codon usage matches the organism's preferred distribution.
CAI-based optimization works, but it is crude. It treats each codon position independently, ignoring the sequence context. It produces repetitive sequences (the same "optimal" codon used everywhere for a given amino acid), which can cause ribosome stalling and mRNA secondary structure problems. And it misses complex dependencies: the optimal codon at position 50 might depend on what codons are at positions 48 and 52, which a frequency table cannot capture.
Our Approach: Masked Language Modeling
We reframe codon optimization as a language modeling problem. Instead of looking up frequencies in a table, we train a transformer on hundreds of thousands of natural coding sequences using masked language modeling (MLM), the same pre-training objective used by BERT, RoBERTa, and Meta's ESM protein models. The model sees a codon sequence with 15% of positions masked and learns to predict the missing codons from context.
What the model learns, implicitly, is the grammar of codon usage: which codon patterns appear in nature, which codons tend to co-occur, and how preferences shift depending on the surrounding sequence context. This is fundamentally richer than a frequency table because the model captures long-range dependencies across the entire coding sequence.
CodonRoBERTa: Our Best Model
After our architecture exploration (see above), CodonRoBERTa-large-v2 emerged as the winner:
# configs/mrna/production/roberta_large_v2.yaml
model_type: roberta
hidden_size: 1024
num_hidden_layers: 24
num_attention_heads: 16
intermediate_size: 4096
vocab_size: 69
max_position_embeddings: 8192
learning_rate: 5e-5 # Critical: lower than v1
warmup_steps: 2000 # Critical: longer warmup
max_steps: 25000Training:
python scripts/training/run_mlm_train.py \
--config configs/mrna/roberta_large_v2.yaml \
--train_file data/mrna/processed/train_250k.fasta \
--output_dir outputs/models/CodonRoBERTa-large-v2Evaluation: Three Metrics That Matter
Evaluating a codon language model is not straightforward. As we learned from the v1/v2 comparison above, a model can have excellent perplexity (accurately predicting masked codons) while having poor biological alignment (predicting codons that nature does not actually prefer). We evaluate on three complementary axes:
1. Perplexity measures how well the model predicts masked codons, computed as the exponentiated cross-entropy loss. A perplexity of 4.10 means the model is, on average, choosing between ~4 equally likely codons at each masked position. Given that most amino acids have 2-6 synonymous codons, this indicates the model has learned meaningful preferences rather than guessing uniformly. Lower is better. CodonRoBERTa-large-v2: 4.10 .
2. CAI Correlation (Spearman) measures whether a model's predicted codon likelihoods align with known biological codon usage preferences. We compute the Codon Adaptation Index for each test sequence, then correlate it with the model's pseudo-log-likelihood score. A positive correlation means the model assigns higher probability to sequences that biology actually uses. This is the metric that matters most for practical codon optimization, because it directly measures whether the model has learned biologically relevant patterns vs. just statistical ones. CodonRoBERTa-large-v2: 0.404 (p < 10^-20).
3. Synonymous Recovery asks: when the model predicts a codon for a masked position, does it at least get the amino acid right? Even if it picks the wrong synonym (e.g., CTT instead of CTC for leucine), predicting the correct amino acid shows the model understands the protein-level constraint. CodonRoBERTa-large-v2: 12.1% top-1 synonymous .
Running the Evaluations
# Perplexity
python scripts/evals/advanced/eval_perplexity.py \
--model outputs/models/CodonRoBERTa-large-v2/final \
--test_file data/mrna/processed/test_6k.fasta \
--output outputs/eval_results/CodonRoBERTa-large-v2/perplexity.json
# CAI Correlation
python scripts/evals/advanced/eval_cai_correlation.py \
--model outputs/models/CodonRoBERTa-large-v2/final \
--test_file data/mrna/processed/test_6k.fasta \
--output outputs/eval_results/CodonRoBERTa-large-v2/cai_correlation.json
# Synonymous Recovery
python scripts/evals/advanced/eval_synonymous_recovery.py \
--model outputs/models/CodonRoBERTa-large-v2/final \
--test_file data/mrna/processed/test_6k.fasta \
--output outputs/eval_results/CodonRoBERTa-large-v2/synonymous_recovery.jsonThe Final Leaderboard
Putting it all together across our model variants:
The RoBERTa family dominates across the board. For production use, CodonRoBERTa-large-v2 is the clear choice: it has the strongest biological alignment (CAI 0.404) while maintaining competitive perplexity. For teams with limited compute, CodonRoBERTa-base delivers nearly the same perplexity at 3.4x fewer parameters. ModernBERT underperformed substantially, which we attribute to its NLP-pretrained weights interfering with codon pattern learning.
Using the Model
from transformers import RobertaForMaskedLM
import torch
# Load model (available soon on Hugging Face)
model = RobertaForMaskedLM.from_pretrained("OpenMed/CodonRoBERTa-large-v2")
tokenizer = CodonTokenizer() # Our custom 69-token vocabulary
# Score a sequence
sequence = "ATG GCT AAA GGT..." # Space-separated codons
inputs = tokenizer(sequence, return_tensors='pt')
with torch.no_grad():
outputs = model(**inputs)
# Pseudo-log-likelihood gives a "naturalness" score
# Predict alternatives for a masked position
masked_seq = "ATG [MASK] AAA GGT..."
inputs = tokenizer(masked_seq, return_tensors='pt')
predictions = model(**inputs).logits
top_codons = predictions[0, mask_pos].topk(5)4. Scaling to Multi-Species
Single-species codon optimization is useful, but limited. Every organism has its own codon usage biases shaped by millions of years of evolution. E. coli favors different codons than human cells, which favor different codons than yeast. A model trained only on E. coli data will not produce optimal codons for human expression.
The industry standard is to use separate CAI tables for each organism. We wanted something better: a single model that understands codon usage across organisms, can be conditioned on a target species, and can transfer knowledge from data-rich organisms (human, with 145k annotated coding sequences) to data-poor ones ( E. coli , with 9k). After establishing CodonRoBERTa-large-v2 as our best architecture on single-species data, we built this system.
The Data Engineering Challenge
Assembling a multi-species codon dataset is not as simple as downloading a few genomes. Each organism lives in a different NCBI RefSeq assembly, with different annotation quality, different CDS boundaries, and different sequence conventions. We wrote an automated pipeline that downloads CDS sequences from 25 organisms, validates them (checking for proper start/stop codons, length divisible by 3, no internal stops), labels each sequence with a species token, and splits into train/test sets with stratification by species.
# scripts/training/download_multispecies_cds.py
# Automated download from NCBI RefSeq for 25 organisms
SPECIES = {
# Bacteria (19 species)
'bacteria': [
('GCF_000005845.2', 'Escherichia coli K-12', 'ECOLI'),
('GCF_000009045.1', 'Bacillus subtilis 168', 'BSUBT'),
('GCF_000006945.2', 'Salmonella enterica', 'SENTE'),
('GCF_000195955.2', 'Mycobacterium tuberculosis', 'MTUBE'),
# ... 15 more bacteria
],
# Yeast (3 species)
'yeast': [
('GCF_000146045.2', 'Saccharomyces cerevisiae S288C', 'YEAST'),
('GCF_000002515.2', 'Schizosaccharomyces pombe', 'SPOMBE'),
('GCF_000027005.1', 'Pichia pastoris', 'PICHIA'),
],
# Mammals (3 species)
'mammals': [
('GCF_000001405.40', 'Homo sapiens GRCh38', 'HUMAN'),
('GCF_000001635.27', 'Mus musculus GRCm39', 'MOUSE'),
('GCF_003668045.3', 'Cricetulus griseus CHO-K1', 'CHO'),
]
}The final dataset spans three domains of biotechnology relevance:
The coverage is deliberate: bacteria are the workhorse of recombinant protein production, yeast dominates industrial biomanufacturing, and mammalian cells (especially CHO and human) are required for therapeutic proteins and mRNA vaccines. These 25 organisms collectively cover the vast majority of real-world codon optimization use cases.
The Tokenization Innovation
A model that sees sequences from 25 different organisms needs to know which organism it is looking at. We solved this by extending our 69-token codon vocabulary with 25 species tokens, creating a 94-token system. Each sequence is prepended with its species token (e.g., [HUMAN] , [ECOLI] , [YEAST] ), so the model learns species-specific codon preferences within a single shared architecture.
# scripts/training/codon_tokenizer.py
class MultiSpeciesCodonTokenizer(CodonTokenizer):
"""Extended tokenizer with species-awareness"""
def __init__(self):
super().__init__()
# 0-4: [PAD], [UNK], [CLS], [SEP], [MASK]
# 5-68: 64 codons (AAA, AAC, ..., TTT)
# 69-93: 25 species tokens
self.species_tokens = [
'[ABAUM]', '[BSUBT]', '[CHO]', '[ECOLI]',
'[HUMAN]', '[MOUSE]', '[YEAST]', # ... +18 more
]
def encode(self, dna_seq: str, species: str = None):
"""Encode with species token prepended"""
ids = super().encode(dna_seq)
if species and species in self.species_to_id:
ids = [self.species_to_id[species]] + ids
return idsThis design has three advantages. First, it enables species-conditioned generation: the same model produces human-optimal or E. coli -optimal codons depending on which species token is prepended. Second, it enables cross-species transfer learning: universal codon patterns (like avoiding certain dinucleotides, or preferring GC-rich codons in GC-rich genomes) are shared across all species, while species-specific preferences are captured by conditioning on the species token. Third, the 94-token vocabulary is backward-compatible with our 69-token single-species models, since the first 69 tokens are identical.
Training the Universal Base Model
The universal base model is a 311.9M-parameter RoBERTa-large, identical in architecture to our single-species v2 but with the expanded 94-token vocabulary. It was trained for 48 hours on 4 A100 GPUs using the full 362k-sequence multispecies dataset.
# configs/mrna/production/roberta_large_multispecies.yaml
model:
name: "CodonRoBERTa-large-multispecies"
vocab_size: 94 # 69 base + 25 species
hidden_size: 1024
num_hidden_layers: 24
num_attention_heads: 16
training:
max_steps: 50000 # ~4.5 epochs over 362k sequences
learning_rate: 5e-5
per_device_train_batch_size: 4 # 4 GPUs = 16 effective
gradient_accumulation_steps: 2 # 32 total batch size
bf16: true
fsdp: "full_shard auto_wrap" # Critical for 311M paramsTraining command:
torchrun --nproc_per_node=4 --master_port=29501 \
scripts/training/run_multispecies_train.py \
--config configs/mrna/production/roberta_large_multispecies.yamlResults:
The test perplexity of 24.9 is higher than our single-species model's 4.01, which might look like a regression. It is not. The multispecies model must learn distinct codon preferences for 25 different organisms, each with its own evolutionary history and tRNA pools. A bacterium like M. tuberculosis (65% GC content) uses completely different codons than human cells (41% GC). The model is solving a fundamentally harder problem, and the perplexity reflects that. What matters is whether species-specific fine-tuning can recover performance, and it does.
Species-Specific Fine-Tuning
The universal base model is a generalist. For production use, specialists perform better. OpenMed's fine-tuning strategy starts from the multispecies checkpoint and trains further on a single species at a lower learning rate (2e-5 vs 5e-5), preserving cross-species knowledge while specializing the model's predictions for one organism.
Dataset splits:
# scripts/training/split_species_datasets.py
# Automated splitting for 6 priority species
RESULTS = {
'HUMAN': {'train': 131_245, 'test': 6_908},
'MOUSE': {'train': 88_022, 'test': 4_633},
'CHO': {'train': 42_541, 'test': 2_239},
'ECOLI': {'train': 8_547, 'test': 450},
'YEAST': {'train': 5_439, 'test': 287},
'PICHIA':{'train': 4_548, 'test': 240},
}Training all three priority species:
# HUMAN: Therapeutic mRNA, vaccines
torchrun --nproc_per_node=4 scripts/training/run_multispecies_train.py \
--config configs/mrna/production/roberta_large_human_finetune.yaml
# ECOLI: Protein expression, metabolic engineering
torchrun --nproc_per_node=4 scripts/training/run_multispecies_train.py \
--config configs/mrna/production/roberta_large_ecoli_finetune.yaml
# CHO: Biopharmaceutical production
torchrun --nproc_per_node=4 scripts/training/run_multispecies_train.py \
--config configs/mrna/production/roberta_large_cho_finetune.yamlComprehensive Results:
The most important result here is the HUMAN model: at 24.3 perplexity, it is the only specialist that beats the universal base, making it our production model for therapeutic mRNA applications. But the ECOLI result is arguably more interesting from a research perspective. With only 8,547 training sequences (compared to 131k for human), the E. coli specialist still improved over the multispecies base. This validates the transfer learning hypothesis: training on 25 species first, then fine-tuning on a small species-specific dataset, works better than training on the small dataset alone. For the many organisms where annotated CDS data is scarce, this approach opens the door to reasonable codon optimization without needing tens of thousands of species-specific sequences.
The CHO model showed slight degradation (25.5 vs 24.9), which we attribute to insufficient training steps. ECOLI got 5,000 steps for 8.5k sequences (0.59 steps per sequence), while CHO got 10,000 steps for 42.5k sequences (0.24 steps per sequence). A rerun with 15,000 steps should close this gap. All three specialists fine-tuned in just 7 hours total, leveraging the 48-hour investment in the multispecies base.
The Complete Model Suite
After 55 hours of training, we have:
All models will be released on Hugging Face under the OpenMed organization. The naming convention follows OpenMed/{model-name} for direct use with from_pretrained() .
Universal Models:
- OpenMed/CodonRoBERTa-large-multispecies (311.9M params) Trained on 25 species
- Perplexity: 24.9
- Use case: Cross-species optimization, rare organisms
Species-Specific Specialists:
- OpenMed/CodonRoBERTa-large-human (311.9M params) Perplexity: 24.3 ( best overall )
- Use case: mRNA vaccines, gene therapy, therapeutic proteins
OpenMed/CodonRoBERTa-large-ecoli (311.9M params)
- Perplexity: 25.3
- Use case: Bacterial protein expression, metabolic engineering
OpenMed/CodonRoBERTa-large-cho (311.9M params)
- Perplexity: 25.5
- Use case: Mammalian cell culture, biopharmaceuticals